臨床試驗係醫療器材產品研發到上市過程中重要的一環。高風險性或新興之醫療器材,須透過臨床試驗以驗證產品效能及安全性,方能通過審查取得上市許可證。本署於96年5月30日公布實施「醫療器材優良臨床試驗基準」,國內執行醫療器材臨床試驗,皆應遵循此項基準,以保障受試者之權益及安全,並確保試驗所得之數據正確可信。
一、辦理查驗登記須於國內進行臨床試驗之品項
已公告需在國內進行臨床試驗之查驗登記醫療器材品項包括隱形眼鏡及其他經中央衛生主管機關指定者。(隱形眼鏡相關減免試驗公告,請參照前行政院衛生署84年9月18日衛署藥字第84061200號、92年3月17日衛署藥字第0920315285號、99年4月27日署授食字第0991400208號公告)
藥商若不確定醫療器材辦理查驗登記時,產品是否須在國內進行臨床試驗,可備齊公告所需資料至本署核辦。(請參照前行政院衛生署92年12月2日衛署藥字第0920332729號函)
二、體外診斷醫療器材(IVD)之臨床評估
體外診斷醫療器材臨床評估非屬醫療法第8 條所稱之人體試驗,符合本部「醫療機構人體試驗委員會得快速審查之案件範圍」,除與公共安全衛生或血液安全相關之新體外診斷醫療器材,必要時本署得要求計畫書送部審查外,其餘計畫書無須送部審查。(請參照前行政院衛生署95年4月21日衛署藥字第0950302084號公告)
三、臨床試驗審理單位
自民國99年1月1日起,所有醫療器材臨床試驗申請案由衛生福利部食品藥物管理署辦理。醫療器材臨床試驗計畫書可平行送醫院人體試驗審議委員會(IRB)及食品藥物管理署審查。
四、臨床試驗申請流程
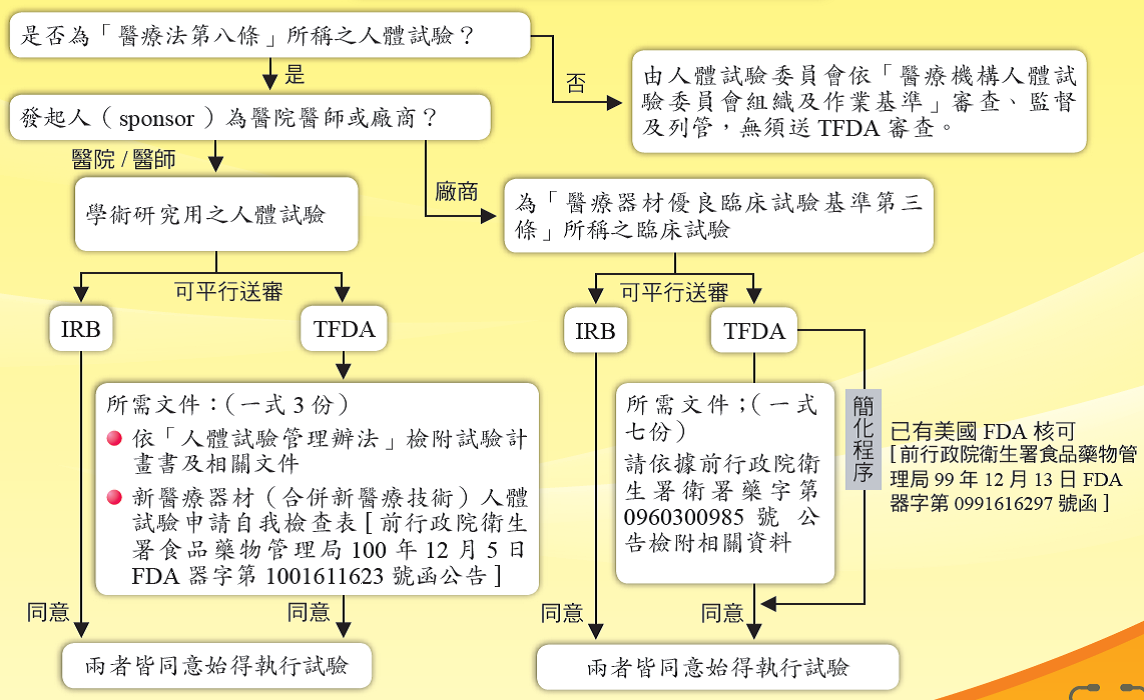
l 前行政院衛生署食品藥物管理局100年12月5日FDA器字第1001611623號函
l 前行政院衛生署96年5月30日衛署藥字第0960300985號公告
五、試驗中所使用之醫療器材
臨床試驗計畫書中所使用醫療器材需於計畫書內容說明試驗中所使用醫療器材之廠牌、規格與數量。若試驗使用對照組產品,需說明對照醫療器材產品之相關訊息。
若使用到未經上市許可之醫療器材應依照專案進口相關規定辦理輸入,所需檢附資料請參考本署網站>業務專區>醫療器材>通關與專案進口。
六、臨床試驗計畫變更
臨床試驗案若有變更須經人體試驗審議委員會(IRB)及食品藥物管理署同意,方可執行變更後內容,但若變更內容未涉及試驗設計與安全性,請於來函載明變更事項(如新增協同主持人、延長受試者收納期間等),則僅需檢送IRB同意函(敘明版本日期)送食品藥物管理署核備,食品藥物管理署將不逐件函覆審查結果。(請參照前行政院衛生署食品藥物管理局99年12月13日FDA器字第0991616851號函)
七、臨床試驗核准後之管理流程
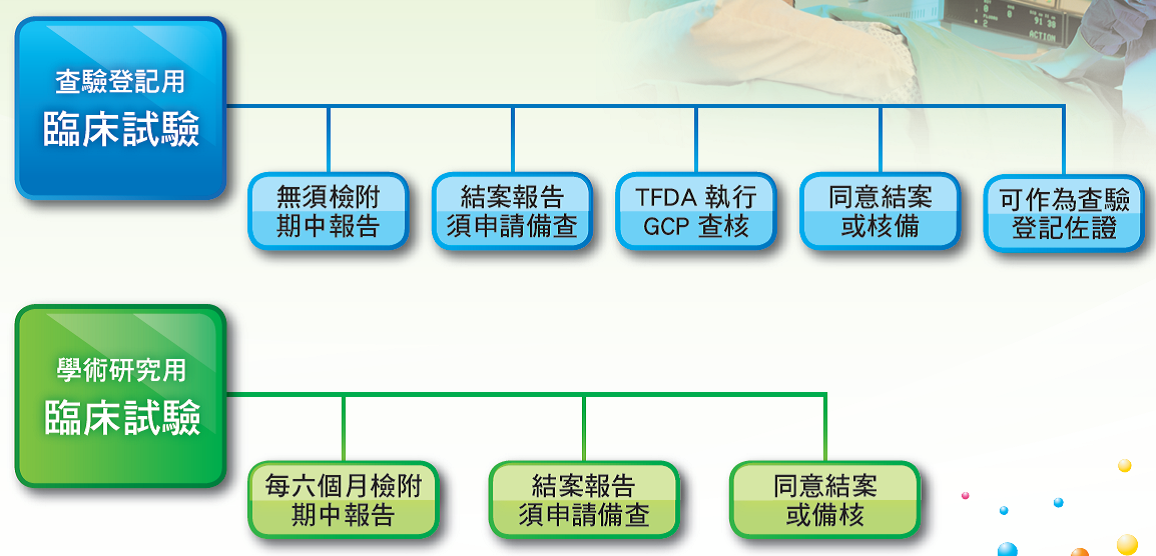
文章來源:衛生福利部食品藥物管理屬